Über Morbus Fabry
Morbus Fabry im Überblick
Was ist Morbus Fabry?
Morbus Fabry gehört zu den sogenannten lysosomalen Speicherkrankheiten, einer Form der Stoffwechselkrankheiten, und wird durch einen Enzymdefekt verursacht. Alternativ wird die Erkrankung auch mit Fabry-Krankheit, Fabry-Anderson-Krankheit oder Fabry-Syndrom bezeichnet.
Morbus Fabry ist erblich bedingt und verläuft bei Männern oft schwerer als bei Frauen. Insgesamt gibt es eine Vielzahl von Symptomen, die unterschiedlich stark ausgeprägt sein können und verschiedene Organe betreffen. Man spricht daher auch von einer Multisystem-Erkrankung. Unbehandelt schreitet die Erkrankung in der Regel immer weiter fort, doch mit der Enzymersatztherapie und der Chaperontherapie stehen den Betroffenen heute wirkungsvolle Behandlungsoptionen zur Verfügung. Wie diese Therapien funktionieren und welche therapeutischen Möglichkeiten außerdem bestehen, lesen Sie im Abschnitt Therapie.1
Wie viele Betroffene gibt es?
Morbus Fabry wird zu den Seltenen Erkrankungen gezählt, das heißt, es sind nicht mehr als fünf von 10.000 Menschen betroffen.2 Tatsächlich wurde zunächst geschätzt, dass nur eine von 40.000–170.000 Personen Morbus Fabry hat.3 In Deutschland haben etwa 1.000 Patient*innen die Diagnose Morbus Fabry erhalten, von denen ca. 800 Betroffene behandelt werden. Es wird jedoch davon ausgegangen, dass es deutlich mehr Menschen gibt, die mit Morbus Fabry leben.4,5 Die Erkrankung wird häufig nicht oder erst spät erkannt, weil die Symptome vielfältig sind und auch Merkmale anderer Krankheitsbilder sein können. Zudem gibt es nur wenige Mediziner*innen, die Erfahrung mit der Diagnostik des Morbus Fabry haben. Doch einige Kliniken haben sich auf die Diagnose und Therapie lysosomaler Speicherkrankheiten spezialisiert und verfügen über die notwendige Expertise; eine Übersicht hierzu finden Sie unter Fachkliniken.
Trotz der Seltenheit der Erkrankung gibt es Selbsthilfeorganisationen, die Ihnen die Möglichkeit geben, mit anderen Betroffenen in Kontakt zu kommen und von einem Erfahrungsaustausch zu profitieren.
Wodurch wird Morbus Fabry verursacht?
Bei Patient*innen mit Morbus Fabry ist ein bestimmter Abschnitt der Erbinformation, das sogenannte GLA-Gen, verändert. Diese Gen-Mutation führt dazu, dass ein Enzym mit dem Namen Alpha-Galaktosidase A (α-GalA) nicht oder nur teilweise funktionsfähig ist. α-GalA ist für den Abbau des Fettstoffs Globotriaosylceramid (Gb3) in den Lysosomen, dem Verdauungsorgan der Zellen, verantwortlich. Aufgrund der mangelnden bzw. fehlenden Aktivität von α-GalA kommt es zu einer fortschreitenden Ansammlung von Gb3 in den Endothelzellen verschiedener Organsysteme. Die genauen Zusammenhänge zwischen der Ablagerung von Gb3 in den Zellen und der Schädigung der betroffenen Organe sind bislang noch nicht endgültig geklärt. Einiges deutet darauf hin, dass verschiedene biochemische Mechanismen, u. a. Entzündungsprozesse, an den Organschädigungen und dem Verlauf der Erkrankung beteiligt sind.1,6
Wie wird Morbus Fabry vererbt?
Erkrankungen, die auf einer Veränderung der Erbinformation beruhen, können vererbt werden. Somit ist Morbus Fabry ein Beispiel für eine Erbkrankheit. Die für die Erkrankung verantwortliche Mutation des GLA-Gens befindet sich auf dem X-Chromosom. Männer haben in ihren Zellen jeweils ein X- und ein Y-Chromosom. Ist das X-Chromosom von der Gen-Mutation betroffen, so erkranken diese Männer an Morbus Fabry. Frauen sind hingegen mit zwei X-Chromosomen ausgestattet, von denen eines nach dem Zufallsprinzip inaktiviert wird. Dass beide X-Chromosomen die Gen-Mutation tragen, ist extrem selten. Häufiger ist nur eines dieser beiden X-Chromosomen betroffen. Wie stark bei einer Frau die Erkrankung ausgeprägt ist, hängt davon ab, ob in den Zellen des jeweiligen Organs in der Mehrzahl intakte oder aber mutierte X-Chromosomen aktiv sind.7
Aufgrund dieses sogenannten X-chromosomalen Erbgangs spielt die Analyse des Familienstammbaums bei der Diagnose von Morbus Fabry sowie die genetische Beratung von Betroffenen und ihren Angehörigen eine wichtige Rolle. Wird bei einem*einer Patient*in Morbus Fabry diagnostiziert, können mithilfe eines Stammbaums weitere Verwandte identifiziert werden, die ebenfalls die Gen-Mutation tragen.
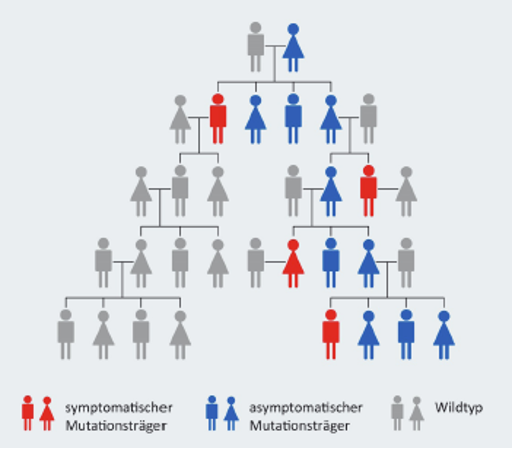
Wie groß die Wahrscheinlichkeit ist, dass die Erkrankung von einem betroffenen Elternteil an dessen Kinder weitergegeben wird, zeigt Abbildung 2.
Welche Formen von Morbus Fabry gibt es?
Klassischer Morbus Fabry
Bei der klassischen Form treten die ersten Symptome in der Regel bereits während der Kindheit auf. Mit zunehmendem Alter weiten sich die Beschwerden auf weitere Organe aus und im Erwachsenenalter kommt es vor allem zu Komplikationen am Herzen, der Niere oder dem Gehirn.3
Atypischer Morbus Fabry
Diese Form wird auch als Late-Onset-Form (englisch: spät beginnend) bezeichnet. Wie der Name andeutet, setzt die Erkrankung später ein als bei der klassischen Form, typischerweise im Alter zwischen 30 und 60 Jahren. Meist ist nur ein Organsystem, überwiegend das Herz, betroffen.7
Weitere Informationen zur klassischen und atypischen Form des Morbus Fabry sind unter Verlauf und Prognose für Sie zusammengestellt.
- Mehta A, Hughes DA. Fabry disease. In: Adam MP, Everman DB, Mirzaa GM, et al. (Hrsg.), GeneReviews(®) [Internet]. University of Washington, Seattle. Copyright © 1993-2022, Seattle (WA), 2002 [Updated 2022]
- Bundesgesundheitsministerium. Seltene Erkrankungen. 2022. https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html, abgerufen am: 28.09.2022
- Paim-Marques L, de Oliveira RJ, Appenzeller S. Multidisciplinary management of fabry disease: current perspectives. J Multidiscip Healthc 2022;15:485-95
- Information der führenden Experten (Key Opinion Leader). 2021. Advisory Board.
- IQVIA TMP Sonderstudie Morbus Fabry. 2020.
- Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30
- Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 2018;123(4):416-27