Diagnose
Diagnose
Wie wird Morbus Fabry festgestellt?
Bei Morbus Fabry handelt es sich um eine seltene Erkrankung, die in der Regel in spezialisierten Fachzentren diagnostiziert und behandelt wird. Zudem kann das Krankheitsbild von Person zu Person stark variieren und umfasst eine ganze Reihe an möglichen Symptomen, die ebenfalls bei anderen – häufigeren – Erkrankungen auftreten. Aus diesen Gründen kann es ab Beginn der Symptome oft einige Jahre (durchschnittlich vier Jahre bei Kindern und 10,5 Jahre bei Erwachsenen) dauern, bis Morbus Fabry diagnostiziert wird.6 Darüber hinaus wird bei einem von vier Fällen zunächst eine falsche Diagnose gestellt.7 Dies kann für die Betroffenen eine schwierige Zeit mit belastenden körperlichen und seelischen Beeinträchtigungen sein.
Morbus Fabry kann sich durch eine Vielzahl unterschiedlicher Krankheitszeichen äußern, die ausführlich unter der Rubrik Symptome beschrieben werden. Doch einige Beschwerden sind besonders häufig und sollten den*die Ärzt*in an diese Stoffwechselkrankheit denken lassen.
Durch Aktivieren des Videos willigen Sie in die Übermittlung Ihrer Daten an Google Ireland Limited sowie gegebenenfalls an ein Drittland ein.
Mehr erfahren
Video laden Youtube immer entsperren
Krankheitszeichen bei Kindern und Jugendlichen1,2
- Kleine rote oder lilafarbene, punktförmige Hautveränderungen, meist im Bereich zwischen dem Bauchnabel und den Oberschenkeln
- Brennende Schmerzen in den Händen und Füßen
- Eingeschränkte oder fehlende Fähigkeit zu Schwitzen
- Beschwerden des Verdauungstraktes wie Durchfall und Bauchschmerzen
- Empfindlichkeit gegenüber Kälte und Hitze
- Starke Müdigkeit und Abgeschlagenheit (Fatigue)
Krankheitszeichen bei Erwachsenen2
- Herzprobleme ohne erkennbare Ursache, z. B. eine Vergrößerung des Herzmuskels der linken Herzkammer oder Herzrhythmusstörungen
- Schlaganfall oder transitorische ischämische Attacke ohne erkennbare Ursache
- Funktionseinschränkung der Niere ohne erkennbare Ursache
Wie wird der Verdacht auf Morbus Fabry bestätigt?
Wurde die Fabry-Krankheit einmal in Betracht gezogen, so ist der endgültige Nachweis recht einfach. Die Diagnostik umfasst zum einen den Enzymaktivitätstest, bei dem die Aktivität des Enzyms Alpha-Galaktosidase A (α-GalA) bestimmt wird. Ist keine oder nur eine minimale Aktivität (< 1 %) feststellbar, handelt es sich um Morbus Fabry der klassischen Form. Bei einer Restaktivität von 1–30 % liegt ein atypischer Morbus Fabry vor.3 Zum anderen wird mithilfe eines Gentests untersucht, ob eine Veränderung des GLA-Gens vorliegt. Welches Nachweisverfahren erforderlich ist, unterscheidet sich bei Männern und Frauen. Die Diagnostik wird heute immer häufiger durch eine Messung der Lyso-Gb3-Menge ergänzt. Lyso-Gb3 ist eine Form des Fettstoffs, der sich aufgrund des Enzymdefekts bei Menschen mit Morbus Fabry ansammelt. Die Bestimmung seiner Konzentration im Blut kann die Erkennung der Erkrankung und nachfolgend die Verlaufsbeobachtung unterstützen.2
Diagnose bei Männern
Bei Männern ist der α-GalA-Aktivitätstest theoretisch ausreichend, um eine sichere Diagnose stellen zu können. Trotzdem wird empfohlen, zusätzlich einen Gentest durchzuführen, um die Art der Mutation des GLA-Gens herauszufinden.4 Zudem kann in Einzelfällen auch bei Männern die Enzymaktivität in der Diagnostik im Normalbereich liegen, obwohl ein Morbus Fabry vorliegt. Auch aus diesem Grund kann ein Gentest sinnvoll sein.
Diagnose bei Frauen
Da bei Frauen die Aktivität von α-GalA im Normalbereich liegen kann, reicht ein Aktivitätstest nicht aus. Vielmehr ist immer eine Analyse des Erbguts notwendig.4
Welche Eingriffe sind für die Diagnosestellung erforderlich?
Sowohl der Aktivitätstest als auch der Gentest und die Messung der Lyso-Gb3-Menge können anhand einer einzigen Blutprobe durchgeführt werden. Besonders schnell und einfach sind sogenannte Trockenbluttests. Hierfür werden wenige Bluttropfen aus der Fingerkuppe auf ein spezielles Filterpapier aufgebracht. Nach der Trocknung wird die Probe an ein Labor verschickt und dort untersucht.5 Solche Trockenbluttests sind für die Patient*innen kostenfrei.
Was passiert nach der Diagnose?
Nachdem die Diagnose Fabry-Krankheit mit den oben beschriebenen Verfahren gestellt wurde, sollte jede betroffene Person umfassend untersucht werden. Dabei sollten alle typischerweise beteiligten Organe berücksichtigt werden, um ein vollständiges Bild des aktuellen Gesundheitszustands zu erhalten. Auf dieser Grundlage wird vom Behandlungsteam gemeinsam mit dem*der Patient*in das weitere therapeutische Vorgehen geplant. Nähere Informationen zu den Behandlungsoptionen sind unter Therapie zusammengefasst.2
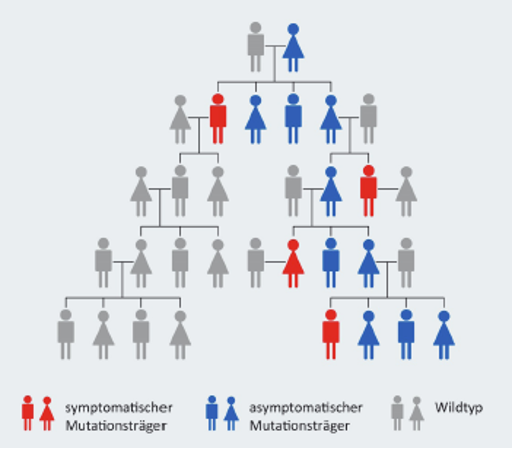
Da es sich bei Morbus Fabry um eine Erbkrankheit handelt, sollte bei neu diagnostizierten Fällen auch immer eine Stammbaumanalyse durchgeführt und das Erbgut der anderen Familienmitglieder untersucht werden. So können weitere Betroffene identifiziert werden und von einer Behandlung profitieren.8
- Germain DP, Fouilhoux A, Decramer S, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clinical Genetics 2019;96(2):107-17
- Mehta A, Hughes DA. Fabry disease. In: Adam MP, Everman DB, Mirzaa GM, et al. (Hrsg.), GeneReviews(®) [Internet]. University of Washington, Seattle. Copyright © 1993-2022, Seattle (WA), 2002 [Updated 2022]
- Paim-Marques L, de Oliveira RJ, Appenzeller S. Multidisciplinary management of fabry disease: current perspectives. J Multidiscip Healthc 2022;15:485-95
- Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 2018;123(4):416-27
- Arndt T. Trockenblut. In: Gressner AM, Arndt T (Hrsg.), Lexikon der Medizinischen Laboratoriumsdiagnostik. Springer Berlin Heidelberg, Berlin, Heidelberg, 2019;2363
- Reisin R, Perrin A, García-Pavía P. Time delays in the diagnosis and treatment of Fabry disease. Int J Clin Pract 2017;71(1)
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004;34(3):236-42
- Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30