Über Epidermolysis bullosa
Epidermolysis bullosa im Überblick
Was ist die Epidermolysis bullosa?
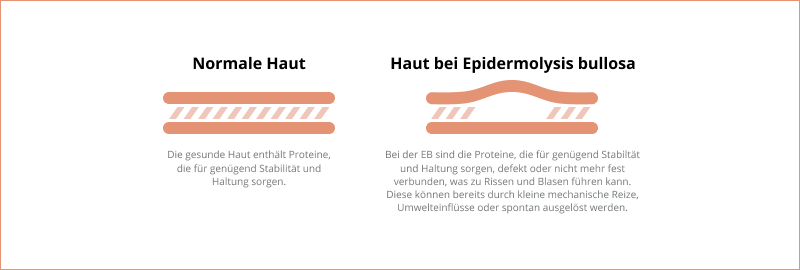
Der Begriff Epidermolysis bullosa (EB) bezeichnet eine Gruppe seltener und unheilbarer Erkrankungen, die durch eine pathologische Verletzlichkeit der Haut gekennzeichnet sind. Die Empfindsamkeit der Haut wird oft mit der Verletzlichkeit von Schmetterlingsflügeln verglichen, weshalb Betroffene auch häufig „Schmetterlingskinder“ genannt werden1.
Bei Menschen mit EB können sich bereits durch leichte mechanische Reize (z. B. Berührungen) oder geringe Umwelteinflüsse Blasen auf der Haut und/oder der Schleimhaut bilden. Wie stark die Symptome sind, hängt unter anderem vom Krankheitstyp und dem jeweiligen Subtyp ab. So kann die Symptomatik auf eine milde, lokal begrenzte Blasenbildung der Haut beschränkt sein oder aber verschiedenste Organe im gesamten Körper betreffen. In diesem Fall spricht man von einer Multisystem-Erkrankung.
In schweren Fällen kann die EB zu Behinderungen führen oder sogar die Lebenszeit verkürzen. Die EB ist eine genetisch bedingte Erkrankung, die meist bereits nach der Geburt oder im Säuglingsalter auftritt. In seltenen Fällen zeigen sich erste Symptome auch erst im fortgeschrittenen Alter2.
Bei der EB werden mehr als 30 Subtypen unterschieden, die in vier Haupttypen unterteilt werden:
- Epidermolysis bullosa simplex
- Junktionale Epidermolysis bullosa
- Dystrophe Epidermolysis bullosa
- Kindler Epidermolysis bullosa
Die verschiedenen Typen werden anhand der zugrundeliegenden genetischen Veränderungen (Genmutation), ihrer Vererbungsmuster, der Schwere der Hautschädigung und des genauen Ortes der Hautschädigung im Bereich der Basalmembranzone unterschieden. Die Basalmembranzone ist der Bereich zwischen der oberen Hautschicht (Epidermis) und der darunterliegenden Bindegewebsschicht (Dermis). Die Basalmembranzone dient als Verankerung zwischen diesen beiden Gewebeschichten und spielt eine wichtige Rolle bei der Stabilität der Haut.
Wie viele Betroffene gibt es?
Die Epidermolysis bullosa (EB) zählt zu den seltenen Erkrankungen und tritt gleich häufig bei Männern und Frauen auf. Zudem ist das Erkrankungsrisiko unabhängig von der ethnischen Zugehörigkeit3. Die Zahl der erkrankten Menschen wird weltweit auf ca. 500.000 geschätzt4.
Einer aktuellen Studie zufolge leben in Deutschland derzeit zwischen 1.800 und 2.000 Menschen mit einer diagnostizierten EB. Es wird allerdings von einer hohen Dunkelziffer ausgegangen, sodass vermutlich sogar 4.000 bis 5.000 Menschen betroffen sind5. Schätzungen zufolge ist eines von 22.000 Neugeborenen in Deutschland von der EB betroffen6.
Wodurch wird die Epidermolysis bullosa verursacht?
Die Haut besteht aus mehreren Schichten, den sogenannten Epithelschichten. Diese bilden gemeinsam das Epithelgewebe, das von speziellen Strukturproteinen zusammengehalten, stabilisiert und an darunterliegende Gewebe verankert wird. Bei Menschen mit Epidermolysis bullosa (EB) führen Genmutationen, also Veränderungen eines Gens in der Erbinformation, zu einer fehlerhaften Herstellung dieser wichtigen Proteine. Infolgedessen verlieren die Hautschichten ihre nötige Stabilität und Haftfähigkeit, was zu einer besonderen Verletzlichkeit führt. Nach aktuellem Stand der Wissenschaft konnten bislang 16 verschiedene Gene identifiziert werden, deren Mutation mit einer EB in Verbindung steht2.
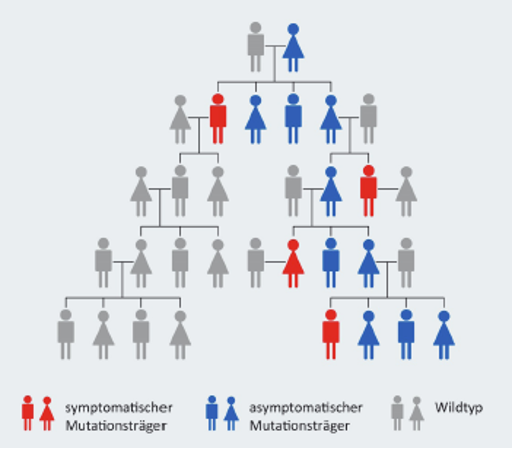
Wie wird die Epidermolysis bullosa vererbt?
Die Epidermolysis bullosa (EB) gehört zu den vererbbaren Erkrankungen. Das bedeutet, dass Betroffene die vorhandene Genmutation in ihrer DNA an ihre Nachkommen weitergeben und diese dann potenziell an EB erkranken können. Die Vererbung der Genmutation kann je nach Erkrankungstyp auf zwei Arten erfolgen:
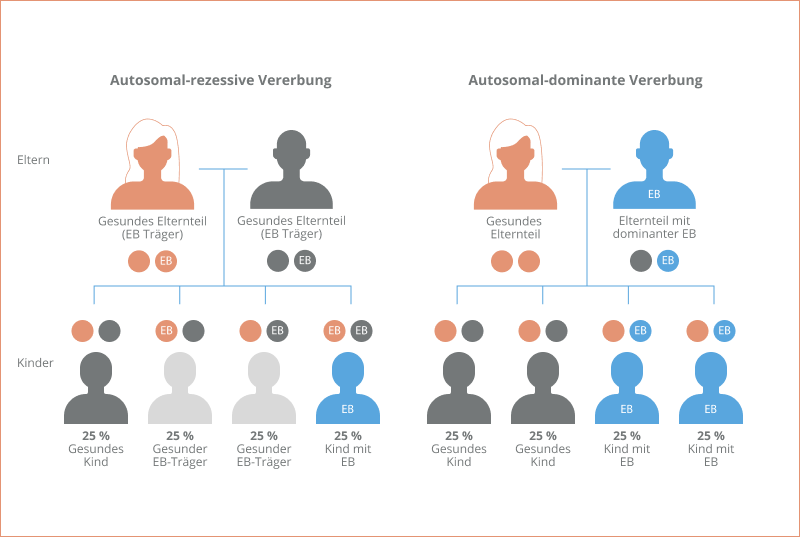
1) Autosomal-rezessiv
Bei dieser Art der Vererbung zeigen beide Elternteile keine Symptome der EB-Erkrankung, sind jedoch Träger*innen der entsprechenden Genmutation. Da jeder Mensch zwei Kopien eines Gens besitzt – je eine auf jedem Chromosom – kann das intakte Gen auf dem anderen Chromosom die Funktion übernehmen und die Bildung funktionsfähiger Proteine ermöglichen, sodass keine Symptome auftreten. In diesem Fall kann die EB nur auftreten, wenn beide Elternteile das Mutation-tragende Gen an ihr Kind weitergeben. Das Risiko, dass ein Kind an EB erkrankt, wenn beide Elternteile Träger*innen eines mutierten Gens sind, liegt statistisch gesehen bei 25 %. Alle vier Krankheitstypen der EB können auf diese Weise vererbt werden1,7.
2) Autosomal-dominant
Während bei allen vier Krankheitstypen ein autosomal-rezessives Vererbungsmuster auftritt, ist bei der EB simplex und der dystrophen EB darüber hinaus auch eine autosomal-dominante Vererbung möglich. Dabei reicht die Weitergabe nur eines mutierten Gens von Mutter oder Vater aus, um eine EB-Erkrankung auszulösen. In diesem Fall kann das nichtmutierte Gen die Funktion des mutierten Gens nicht übernehmen. Das Risiko, dass unter diesen Umständen ein Kind an EB erkrankt, liegt statistisch gesehen bei 50 %1,7.
In seltenen Fällen kann es auch vorkommen, dass die EB erstmalig und spontan bei einem Kind auftritt, obwohl die Eltern keine entsprechende Mutation aufweisen. Die Wahrscheinlichkeit, dass das Kind diese neu aufgetretene EB-Mutation weitervererbt, liegt jedoch bei 50 %, da sie einem ausschließlich dominanten Erbgang folgt1.
Wie unterscheiden sich die vier Krankheitstypen der Epidermolysis bullosa?
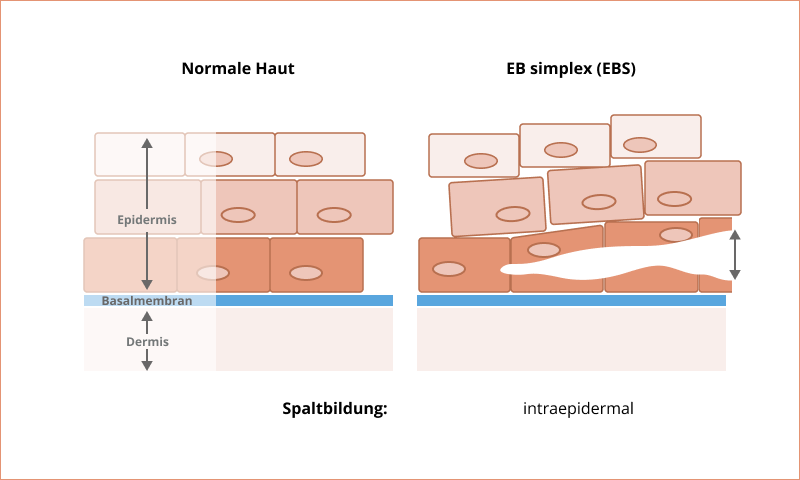
Epidermolysis bullosa simplex
Epidermolysis bullosa simplex (EBS) ist der häufigste EB-Krankheitstyp und macht ungefähr 70 % aller Krankheitsfälle aus. Die Blasenbildung bei der EBS erfolgt vor allem in den höheren Hautschichten oder in den auf der Basalmembran aufliegenden Keratinozyten. Dabei handelt es sich um spezielle Zellen, die für die Produktion von Keratin (Hornsubstanz) zuständig sind. Die EBS wird bis auf wenige Ausnahmen autosomal-dominant vererbt. Nur in ca. 5 % der Fälle liegt ein autosomal-rezessives Vererbungsmuster vor2.
Die EBS geht mit vielen Subtypen und einer großen Vielfalt an Schweregraden einher7. Diese reichen von geringfügiger Blasenbildung an den Händen und Füßen bis hin zu Typen, bei denen es unter anderem zu Erkrankungen des Herzens (z. B. der Herzmuskulatur) und der Nieren kommen kann9,10. Schwere Ausprägungen der EBS können zudem die Lebenserwartung verringern9.
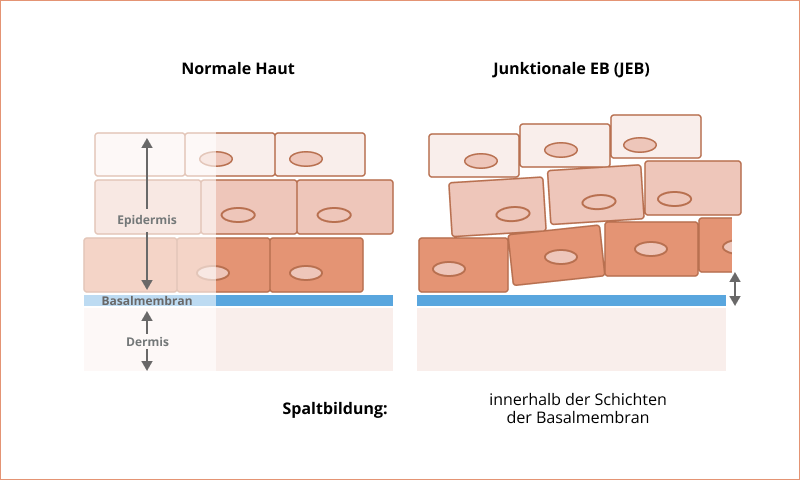
Junktionale Epidermolysis bullosa
Bei der junktionalen Epidermolysis bullosa (JEB) werden Blasen an der Basalmembran zwischen der oberen Hautschicht (Epidermis) und der darunterliegenden Bindegewebsschicht (Dermis) gebildet. Die JEB macht ungefähr 5 % aller EB-Fälle aus und wird autosomal-rezessiv vererbt2.
Zu den häufigsten JEB-Subtypen zählen der intermediäre und der schwere Typ9. Charakteristisch für alle JEB-Subtypen sind Veränderungen der Nagelform, -farbe und -struktur sowie des Nagelwachstums. Darüber hinaus kann es zu einem Verlust der Nägel kommen. Ansonsten ist die Symptomatik der JEB-Subtypen sehr variabel2,8.
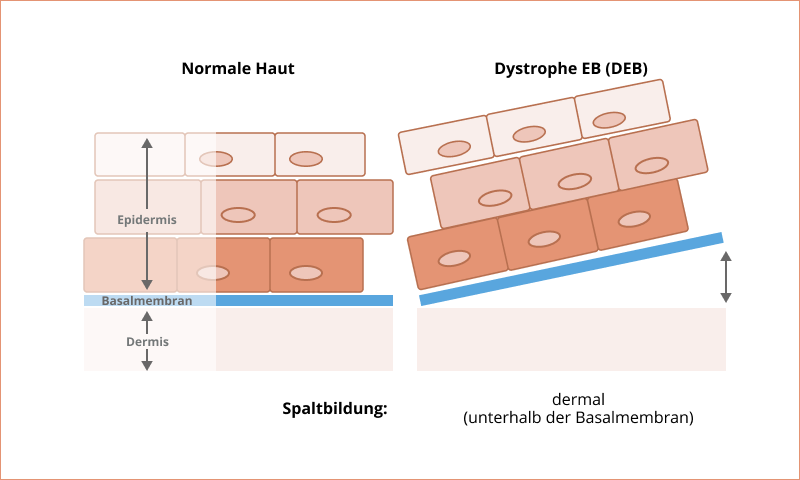
Dystrophe Epidermolysis bullosa
Menschen mit dystropher Epidermolysis bullosa (DEB) zeigen eine Blasenbildung unterhalb der Basalmembran. Insgesamt weisen ca. 25 % der EB-Betroffenen eine DEB auf. Abhängig vom Subtyp erfolgt die Vererbung autosomal-rezessiv oder autosomal-dominant, wobei die rezessiv vererbten Typen meistens schwerwiegendere Krankheitsverläufe zur Folge haben2,9.
Bei mittelschweren bis schweren Subtypen ist im Lebensverlauf die Entwicklung von bösartigem Hautkrebs, sogenannter Plattenepithelkarzinome, möglich12.
Kindler Epidermolysis bullosa
Anders als bei den anderen Typen der Epidermolysis bullosa (EB) ist die Blasenbildung bei der Kindler Epidermolysis bullosa (KEB) nicht auf einen Hautbereich beschränkt. Mit etwas mehr als 250 Fällen weltweit ist die KEB der seltenste Typ der EB. Die Vererbung folgt einem autosomal-rezessiven Muster.
Die Erkrankung zeigt sich besonders durch Blasenbildung an Händen und Füßen, eine hohe Lichtempfindlichkeit und eine ungleichmäßige Ablagerung von Pigmenten in der Haut2.
Weitere Informationen zu den vier Krankheitstypen der Epidermolysis bullosa sind unter Symptome und Verlauf und Prognose zusammengestellt.
- DEBRA. Was ist EB? Epidermolysis Bullosa Infografiken. 2024. https://www.ieb-debra.de/wp-content/uploads/Was-ist-Epidermolysis-bullosa-EB.pdf, abgerufen am: 26.07.2024
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers 2020;6(1):78
- Dorn B. Epidermolysis bullosa (Bachelorarbeit an der Medizinischen Universität Graz). 2014. https://online.medunigraz.at/mug_online/wbAbs.getDocument?pThesisNr=45212&pAutorNr=&pOrgNr=1, abgerufen am: 19.07.2024
- Baardman R, Yenamandra VK, Duipmans JC, et al. Novel insights into the epidemiology of epidermolysis bullosa (EB) from the Dutch EB Registry: EB more common than previously assumed? J Eur Acad Dermatol Venereol 2021;35(4):995-1006
- Has, Cristina et al. JEADV vol. 37,2 (2023): 402-410
- Has C, Hess M, Anemüller W, et al. Epidemiology of inherited epidermolysis bullosa in Germany. J Eur Acad Dermatol Venereol 2023;37(2):402-10
- EB-Haus Austria. EB-Handbuch. Genetik und Vererbung. 2015. https://www.eb-haus.org/fileadmin/user_upload/Media_Library/EB-Handbuch/PDFs_Deutsch/Genetik.pdf, abgerufen am: 19.07.2024
- Mariath LM, Santin JT, Schuler-Faccini L, et al. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol 2020;95(5):551-69
- Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol 2020;183(4):614-27
- Sánchez-Jimeno C, Escámez MJ, Ayuso C, et al. Genetic diagnosis of epidermolysis bullosa: recommendations from an expert Spanish research group. Actas Dermosifiliogr (Engl Ed) 2018;109(2):104-22
- UK-Freiburg. Pathogenese und Klassifikation der Epidermolysis bullosa. 2018. https://www.uniklinik-freiburg.de/hautklinik/kompetenzzentrum-fuer-fragile-haut-und-epidermolysis-bullosa/epidermolysis-bullosa-zentrum/pathogenese-klassifikation.html, abgerufen
- Has C, Fischer J. Epidermolysis bullosa hereditaria. Medizinische Genetik 2019; https://doi.org/10.1007/s11825-019-00266-3