Über Alpha-Mannosidose
Alpha-Mannosidose im Überblick
Was ist Alpha-Mannosidose?
Alpha-Mannosidose ist eine genetisch bedingte Erkrankung aus der Gruppe der sogenannten lysosomalen Speichererkrankungen. Sie gehört zu den seltenen Erkrankungen und tritt bei ca. 1-2 von 1.000.000 Menschen auf. Verursacht wird sie durch verschiedene Veränderungen, sogenannten Mutationen, in den Abschnitten des Erbmaterials, die für die Ausprägung des lysosomalen Enzyms Alpha-Mannosidase verantwortlich sind.
Früher wurde die Alpha-Mannosidose in drei Typen unterteilt: leichte Form, mittelschwere Form und schwere Form. Da die einzelnen Formen fließend ineinander übergehen können, ist diese strenge Einteilung inzwischen allerdings nicht mehr üblich.
Das klinische Bild der Alpha-Mannosidose ist insgesamt individuell sehr variabel. Es handelt sich aber stets um eine progressive, also voranschreitende Erkrankung mit großem Einfluss auf die Lebensqualität, da im Krankheitsverlauf meist das Skelett, das Gehör, das Nervensystem und das Immunsystem der Patient*innen betroffen sind.3
Informativer Kurzfilm zur Alpha-Mannosidose
In diesem kurzen Film finden Sie umfassende Informationen zur Diagnostik, zu den wichtigsten Symptomen und zur Therapie der Alpha-Mannosidose. Frau Prof. Dr. Julia B. Hennermann, die sich auf pädiatrische Stoffwechselerkrankungen spezialisiert hat und Prof. Dr. Dag Malm, der maßgeblich zur Erforschung dieser lysosomalen Speichererkrankungen beigetragen hat und selbst Vater von zwei erkrankten Töchtern ist, vermitteln Ihnen hier ein umfassendes Bild.
Ursachen und Vererbungsmuster
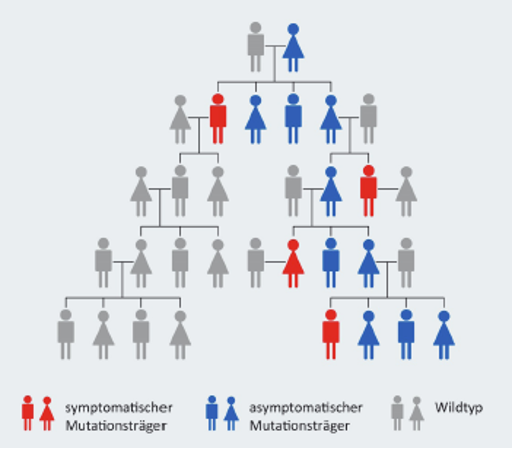
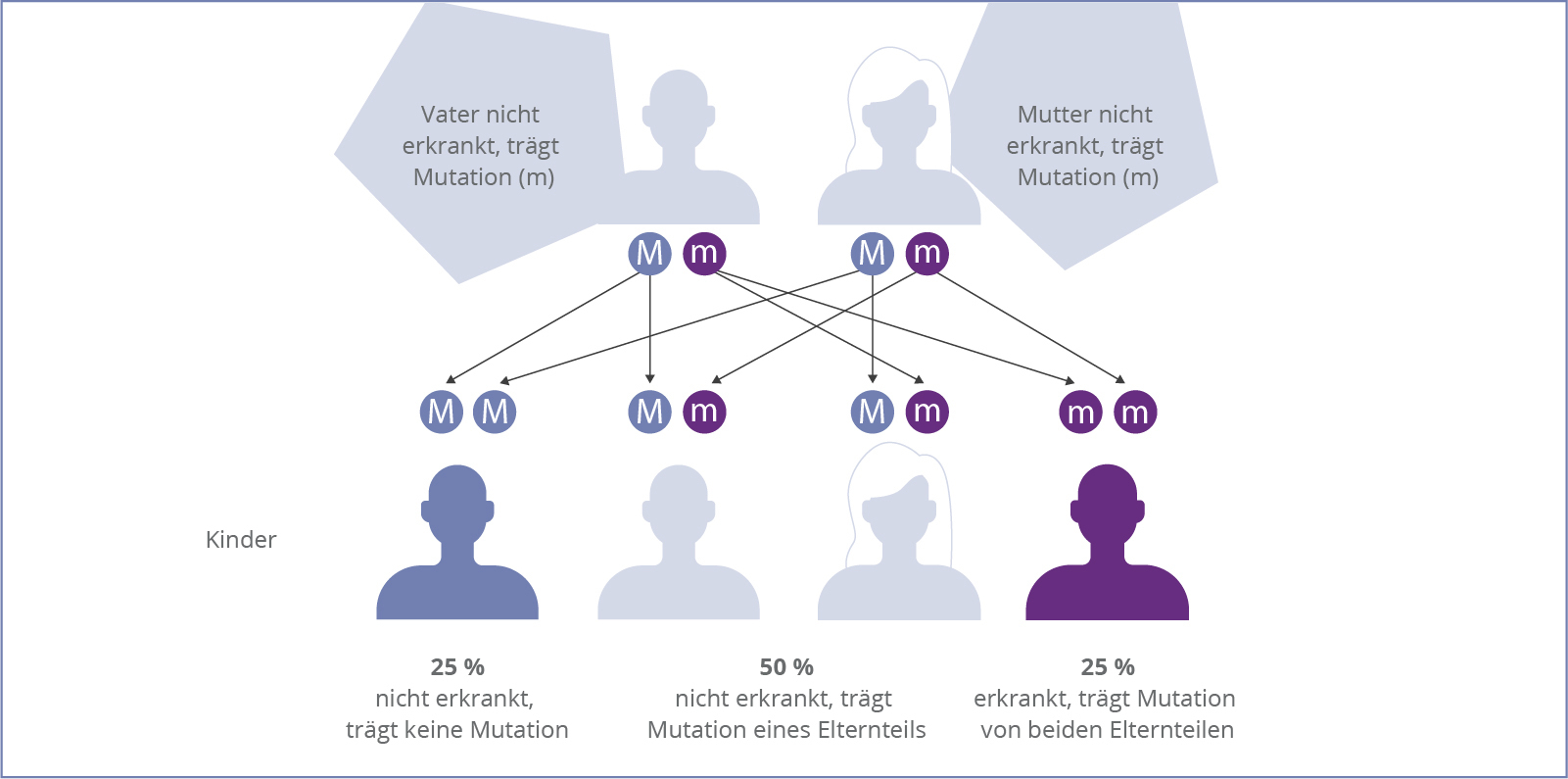
Die Alpha-Mannosidose ist eine autosomal-rezessiv vererbte Krankheit, d.h. die Erkrankung tritt nur dann in Erscheinung, wenn sich auf den jeweiligen Chromosomen beider Elternteile eine krankmachende Veränderung befindet. Durch diesen Defekt wird der Abbau von mannosehaltigen Mehrfachzuckern gestört, sogenannter Oligosacchariden.
Da es eine Vielzahl möglicher Mutationen gibt, ist es für ein Paar, das bereits ein Kind mit Alpha-Mannosidose hat, ratsam, sich bezüglich der weiteren Familienplanung an eine genetische Beratung zu wenden. So kann ein besseres Verständnis der Betroffenen gegenüber der Erkrankung geschaffen werden.9
Was passiert bei Alpha-Mannosidose im Körper?
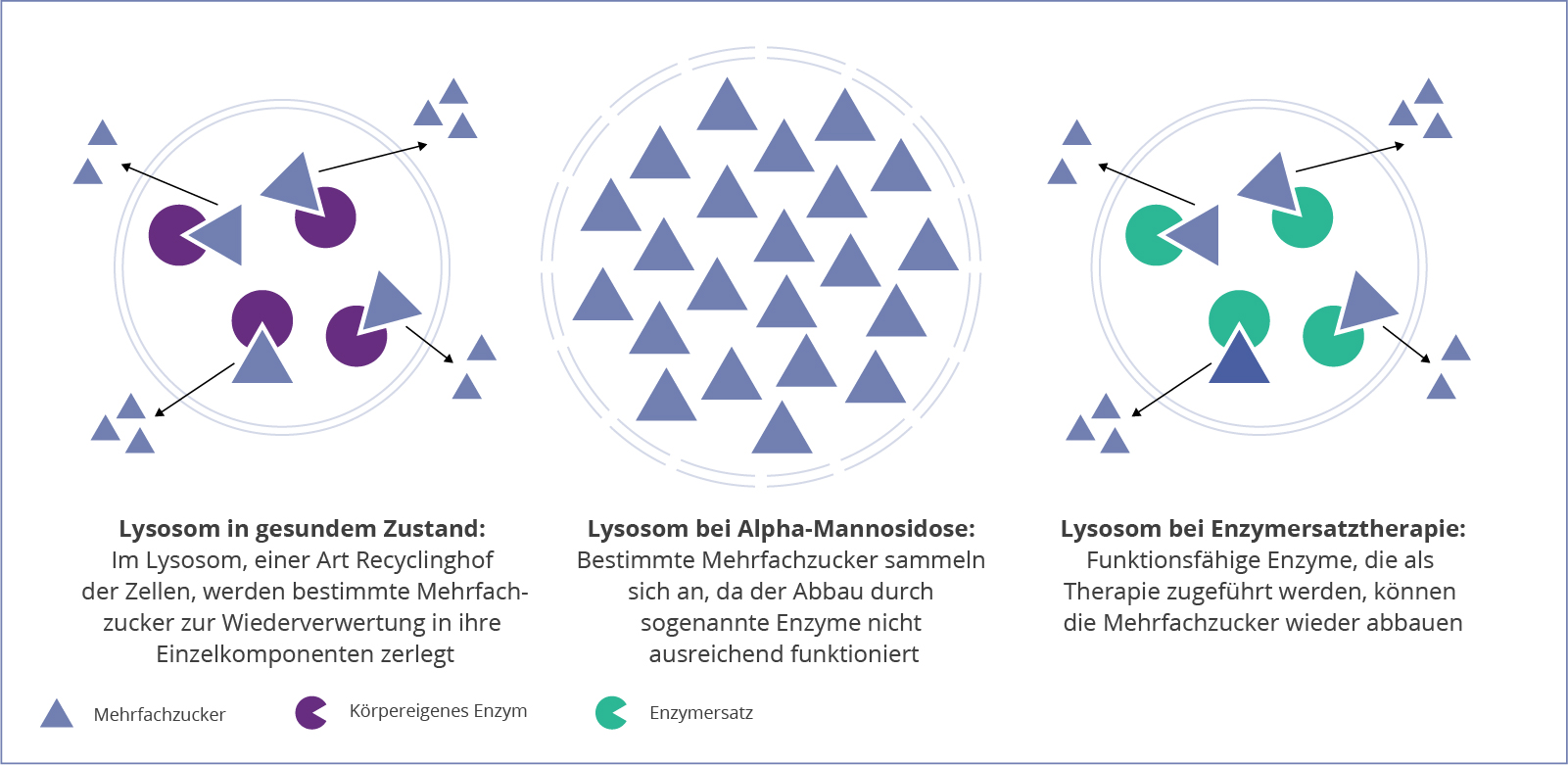
Als lysosomale Speichererkrankung steht die Alpha-Mannosidose mit der Ansammlung einer Zuckerart, der Mannose, in den Zellen in Zusammenhang. Die Erkrankung nimmt dabei ihren Anfang in den Lysosomen, den “Recyclinghöfen” der Zellen. Durch einen Defekt im Gen MAN2B1, wird ein bestimmtes Enzym nicht erfolgreich gebildet. Diese Glykosidase ist normalerweise für den Abbau von hybriden und komplexen mannosereichen Oligosacchariden im Lysosom verantwortlich.1,3
Der Auf- und Abbau der Oligosaccharide ist ein entscheidender Mechanismus für verschiedene Bereiche des Körpers. Diese komplexen Zucker sind unter anderem beim Aufbau von Knochen, Knorpel, Sehnen oder Haut beteiligt. Dabei findet in den Lysosomen ein ständiges Recycling dieser Moleküle statt, wobei neue Oligosaccharide produziert und alte abgebaut werden. Durch einen Funktionsverlust oder das Fehlen der Alpha-Mannosidase, sammeln sich die Oligosaccharide mit der Zeit in den Lysosomen an, was zur Schädigung und letztlich zum Absterben der Zellen führen kann.1,3
Verlauf und Therapie der Alpha-Mannosidose
Das klinische Bild der Alpha-Mannosidose kann sich im Krankheitsverlauf ändern. Unter Verlauf und Prognose finden Sie mehr Informationen über die fortschreitende Erkrankung in den verschiedenen Lebensphasen. Für die Alpha-Mannosidose stehen inzwischen Therapiemöglichkeiten zur Verfügung, die von einer ursächlichen Enzymersatztherapie über eine symptomatische Behandlung bis hin zur Unterstützung durch Hilfsmittel wie etwa Gehhilfen reichen.2,10 Möchten Sie mehr zur Therapie der Alpha-Mannosidose erfahren, folgen Sie diesem Link.
- Beck M, et al. Orphanet J Rare Dis. 2013;8:88.
- Malm D, Nilssen Ø. Orphanet J Rare Dis. 2008;3:21.
- Borgwardt L et al. Orphanet J Rare Dis. 2015;10:70.
- Neufeld EF, et al. The Metabolic and Molecular Bases of Inherited Disease. 2001;3421–3452.
- Grosse H. Pädiatrie. 2018;30,30.
- Beck, M. Dtsch Arztebl. 2001;98:34-35.
- Alpha-Mannosidosis Mutation Database. Tromsoe University. https://apex.jupiter.no/apex/f?p=101:1 (zuletzt aufgerufen: April 2022).
- Ceccarini MR, et al. Int J Mol Sci. 2018;19:1500.
- Guide to understanding mannosidosis. Society for Mucopolysaccharide Diseases. http://www.mpssociety.org.uk/wp-content/uploads/2016/07/guide-alphamannosidosis-2013.pdf (zuletzt aufgerufen: April 2022).
- Fachinformation Lamzede®