Über Beta-Thalassämie
Beta-Thalassämie im Überblick
Was genau sind Beta-Thalassämien?
Die Beta-Thalassämie – auch als Cooley’s Anämie bekannt – ist eine seltene, angeborene Blutkrankheit. Sie entsteht durch eine Veränderung (Mutation) in einem bestimmten Gen, das für die Herstellung von Hämoglobin verantwortlich ist. Hämoglobin ist ein lebenswichtiger Bestandteil unserer roten Blutkörperchen – es sorgt dafür, dass Sauerstoff im Körper transportiert wird.1
Bei Menschen mit Beta-Thalassämie wird Hämoglobin nicht richtig gebildet. Dadurch sind die roten Blutkörperchen oft zu klein, zu wenige und enthalten weniger Hämoglobin als nötig. Die Folge: Der Körper bekommt nicht genug Sauerstoff – das kann zu Müdigkeit, Schwäche und anderen Beschwerden führen.1
Das Wort „Thalassämie“ stammt aus dem Griechischen: thalassa bedeutet „Meer“ und aemia steht für „Blut“. Der Name wurde gewählt, weil die Krankheit ursprünglich vor allem in Ländern rund ums Mittelmeer vorkam – zum Beispiel in Griechenland oder Italien. Sie ist aber auch in Teilen Asiens, des Nahen Ostens und Afrikas verbreitet. Heute trifft man die Krankheit weltweit an – auch in Deutschland, vor allem durch Migration.1
In Deutschland leben schätzungsweise etwa 500 Menschen mit der schweren Form, der sogenannten Thalassaemia major. Weitere rund 160.000 Menschen tragen die milde Form, Thalassaemia minor. Diese Menschen haben meist keine oder nur sehr leichte Symptome, können die Genveränderung aber an ihre Kinder weitergeben.1
Wie entsteht Beta-Thalassämie?
Die Beta-Thalassämie entsteht durch eine Veränderung in den Erbanlagen (einen sogenannten Gendefekt). Diese Veränderung sorgt dafür, dass der Körper ein wichtiges Eiweiß – das sogenannte Beta-Globin – nicht richtig herstellen kann. Beta-Globin ist ein Teil des roten Blutfarbstoffs Hämoglobin, der dafür zuständig ist, Sauerstoff im Körper zu transportieren.1
Hämoglobin besteht normalerweise aus vier Eiweiß-Bausteinen – zwei Alpha-Ketten und zwei Beta-Ketten. Wenn die Beta-Ketten nicht richtig gebildet werden, funktioniert das Hämoglobin nicht mehr so, wie es sollte. Das hat Folgen:1
- Es entstehen weniger rote Blutkörperchen.
- Die roten Blutkörperchen sind kleiner als normal.
- Sie können weniger Sauerstoff transportieren.
Das führt dazu, dass der Körper schlechter mit Sauerstoff versorgt wird – was sich oft durch Müdigkeit, Blässe, Kurzatmigkeit oder allgemeine Schwäche bemerkbar macht.1

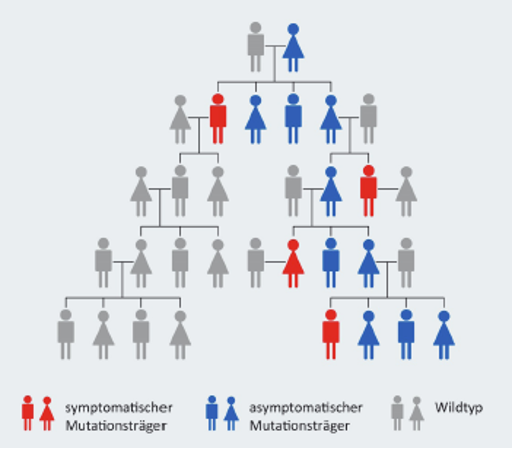
Beta-Thalassämie ist eine erblich bedingte Krankheit, das heißt: Sie wird von den Eltern an ihre Kinder weitergegeben. Die Vererbung erfolgt auf eine bestimmte Weise, die man „autosomal-rezessiv“ nennt.1
Was bedeutet das?
- Autosomal heißt: Das betroffene Gen liegt nicht auf einem Geschlechtschromosom. Deswegen können Mädchen und Jungen gleichermaßen betroffen sein.
- Rezessiv heißt: Ein Kind bekommt die Krankheit nur dann vollständig, wenn es das defekte Gen von beiden Elternteilen erbt – also von der Mutter und vom Vater. Wenn nur ein Elternteil das veränderte Gen vererbt, hat das Kind in der Regel keine Beschwerden, kann das Gen aber weitervererben – man spricht dann von einem sogenannten „Anlageträger“ (auch „Thalassaemia minor“ genannt)
Was ist Hämoglobin – und warum ist es so wichtig?
Hämoglobin ist der rote Blutfarbstoff in unseren roten Blutkörperchen. Es handelt sich um ein spezielles Eiweiß, das eine zentrale Aufgabe hat: Es transportiert Sauerstoff von der Lunge in den ganzen Körper und bringt das verbrauchte Kohlendioxid zurück zur Lunge.
Wie ist Hämoglobin aufgebaut?
Hämoglobin besteht aus zwei Hauptbestandteilen:1
- Globinketten: Das sind Eiweißbausteine, die Struktur und Funktion des Hämoglobins bestimmen. Es gibt fünf Arten davon: Alpha, Beta, Gamma, Delta und Epsilon. Ein Hämoglobin-Molekül besteht aus vier Globinketten – immer zwei gleiche Paare.
- Häm-Eisen: In jede dieser Globinketten ist ein eisenhaltiges Molekül eingebettet. Man nennt es Häm-Eisen und es ist entscheidend dafür, dass Sauerstoff gebunden und wieder abgegeben werden kann. Pro Hämoglobin-Molekül gibt es vier Häm-Eisen – also vier Sauerstoffplätze.
Welche Hämoglobin-Typen gibt es?
Im Laufe des Lebens gibt es verschiedene Formen von Hämoglobin – zwei davon sind besonders wichtig im Zusammenhang mit Beta-Thalassämie:2
- Fetales Hämoglobin (HbF): Es kommt vor allem vor der Geburt vor und besteht aus zwei Alpha- und zwei Gamma-Ketten. Nach der Geburt wird es zunehmend durch HbA ersetzt.
- Erwachsenen-Hämoglobin (HbA): Es besteht aus zwei Alpha- und zwei Beta-Ketten und übernimmt nach der Geburt die Hauptrolle im Sauerstofftransport.

Was passiert bei Beta-Thalassämie im Körper?
Beta-Thalassämie ist eine genetische Erkrankung, bei der das Erbgut für die Beta-Ketten im Hämoglobin defekt ist. Das führt zu zwei großen Problemen:
Zu wenig Beta-Ketten – zu wenig Hämoglobin
- Der Körper kann nicht genug HbA bilden.
- Es entstehen weniger und kleinere rote Blutkörperchen, die weniger Sauerstoff transportieren können.
- Die Folge: es entwickelt sich eine Blutarmut (Anämie) mit typischen Symptomen.
Überschuss an Alpha-Ketten – instabile Blutzellen
- Die Alpha-Ketten werden weiter produziert, aber wegen dem Mangel an Beta-Ketten können nur wenige HbA-Moleküle gebildet werden.
- Die überschüssigen Alpha-Ketten zerfallen schon im Knochenmark.
- Die roten Blutzellen sind dadurch instabil und werden vom Körper schnell wieder abgebaut – noch bevor sie richtig arbeiten können.
Formen der Beta-Thalassämie
Beta-Thalassämie tritt in drei verschiedenen Ausprägungen auf – je nachdem, wie stark die Bildung des roten Blutfarbstoffs gestört ist. Entscheidend ist dabei, wie viele defekte Gene vererbt wurden und wie schwerwiegend der Defekt der Gene ist.
1. Thalassaemia major – die schwere Form1
- Diese Form entsteht, wenn beide Elternteile das defekte Gen weitergeben. Man spricht dann von einer homozygoten Erkrankung.
- Ohne Behandlung ist Thalassaemia major lebensbedrohlich.
- Die Krankheit zeigt sich bereits im Säuglingsalter – mit starker Blutarmut, Wachstumsstörungen und Organschäden.
- Betroffene sind auf regelmäßige Bluttransfusionen und ärztliche Betreuung angewiesen.
2. Thalassaemia intermedia – die mittelschwere Form1
- Hier liegt entweder bei beiden Eltern weniger schwerer Defekt des betroffenen Gens vor oder jedes Elternteil hat ein unterschiedlich defektes Gen.
- Das bedeutet: Die Bildung von Hämoglobin ist zwar eingeschränkt, funktioniert aber noch teilweise.
- Die Symptome können sehr unterschiedlich ausgeprägt sein: bei einigen bereits im Kindesalter, bei anderen erst im Erwachsenenalter.
- Nicht alle Betroffenen brauchen Transfusionen – manche kommen jahrelang ohne Behandlung aus, andere benötigen gezielte medizinische Unterstützung.
3. Thalassaemia minor – die milde Form1
- Hier wird nur von einem Elternteil ein defektes Gen weitergegeben.
- Das gesunde Gen von dem anderen Elternteil reicht meist aus, um genügend HbA-Hämoglobin zu bilden.
- Diese Form verläuft oft beschwerdefrei.
- Viele Menschen wissen gar nicht, dass sie Träger*innen des Gens sind.
- Wichtig: Menschen mit Thalassaemia minor können ein defektes Gen an ihre Kinder weitergegeben werden. Deshalb ist ein Gentest bei der Familienplanung sinnvoll.
Was bei Beta-Thalassämie noch wichtig ist
Auf dieser Seite finden Sie noch weitere hilfreiche Informationen zum Thema Beta-Thalassämie. Erfahren Sie zum Beispiel wichtiges zur Diagnose der BT, welche Symptome bei BT auftreten können oder welche Therapieoptionen infrage kommen.
- Cario H. Beta (ß) –Thalassämie. 2023. online verfügbar unter: https://www.kinderblutkrankheiten.de/sites/gpoh/kinderkrebsinfo/kinderblutkrankheiten/content/e97222/e96941/e96942/e104986/e106654/ss_Thalassaemie.pdf (Zuletzt aufgerufen März 2025).
- Thieme via medici. Hämoglobin Synthese und Aufbau. 2025. Online verfügbar unter https://viamedici.thieme.de/lernmodul/549519/539519/ (Zuletzt aufgerufen März 2025).